
The directed differentiation of human embryonic stem cells (hESCs) or human induced pluripotent stem cells (hiPSCs) into hepatocytes could facilitate a rational study of the molecular mechanisms underlying human liver development as well as provide a renewable source of exogenous hepatocytes for drug toxicity testing and cell-based therapeutics. Moreover, if hepatocytes were produced from hiPSCs originating from patients with inborn errors of hepatic metabolism, such cells could be used for modeling liver disease. Here we present a step-wise protocol for efficiently and reproducibly inducing the differentiation of hepatocytes from human pluripotent stem cells under highly defined conditions.
1. Introduction
Hepatocytes contribute ∼70% of hepatic mass and are responsible for the majority of functions associated with the liver including detoxification, carbohydrate metabolism, protein storage, urea synthesis, formation of bile, cholesterol homeostasis and the secretion of a plethora of proteins that are present within serum. Studies in mice and rats have demonstrated that transplant of exogenous hepatocytes can successfully replace dysfunctional host parenchymal cells (Demetriou et al., 1986b; Demetriou et al., 1986a; Overturf et al., 1996). Such studies have led to clinical trials to test the feasibility of correcting inborn errors of hepatic metabolism using exogenous cell transplant (Grompe, 2006). Although preliminary results from such studies are encouraging, the availability of donor livers is limited raising the need for an alternative source of human hepatocytes.
One possible source of hepatocytes could be provided by human pluripotent stem cells because such cells have a high capacity for self-renewal as well as the potential to differentiate into a wide variety of somatic cell types including hepatocytes (Thomson et al., 1998; Yu et al., 2007; Takahashi et al., 2007). The generation of hepatocytes from human induced pluripotent stem cells (hiPSCs) is particularly appealing because it could be used to model heritable hepatic disorders such as familial hypercholesterolemia and alpha-1-antitrypsin deficiency (Rashid et al., 2010).
A large number of groups have described protocols that can induce the differentiation of cells with gene expression and functional profiles that closely resemble that of hepatocytes using both hESCs and hiPSCs (Rambhatla et al., 2003; Cai et al., 2007; Agarwal et al., 2008; Hay et al., 2008; Shiraki et al., 2008; Cho et al., 2008; Basma et al., 2009; Song et al., 2009; Si-Tayeb et al., 2010b; Sullivan et al., 2010). Most of the approaches that have been described have successfully exploited data generated from decades of research into the molecular mechanisms that control hepatocyte differentiation during embryogenesis (Si-Tayeb et al., 2010a; Zaret, 2008). In addition, unlike many other cell fates, human pluripotent stem cells seem particularly amenable to following a hepatic fate. While the results have generally been impressive, it is important to keep in mind that no protocol has successfully generated hepatocytes that can fully replace the parenchymal cells of the mouse liver in the manner that primary human hepatocytes are capable of (Azuma et al., 2007). Although such a caveat may impact the use of such cells for cell transplant, hESC– and hiPSC–derived hepatocytes have already proven to be a valuable tool to study human hepatocyte dysfunction and differentiation and are likely to provide platforms for drug discovery in the near future (Yusa et al., 2011; Rashid et al., 2010; Delaforest et al., 2011).
Original protocols, which were adapted from studies using mouse ES cells, generally used suspension culture of stem cell aggregates (embryoid bodies) as a mechanism to initiate differentiation. Although effective in inducing differentiation, the procedure tends to be chaotic and as a consequence generates multiple cell lineages that commonly require subsequent cell sorting or selection to be useful. Moreover, the efficacy of producing high quality hepatocytes through an embryoid body intermediate tends to be variable, which affects reproducibility and complicates interpretation of data. Similar problems can be associated with protocols that include culture of differentiated cells using feeder layers or include ill-defined additives such as fetal bovine serum that typically exhibit substantial lot–to–lot variation. With this in mind we have described a protocol, illustrated in Figure 1, that relies on highly defined culture conditions that facilitate the reproducible differentiation of hepatocytes from a wide-range of hESCs and hiPSCs (Delaforest et al., 2011; Si-Tayeb et al., 2010b). Although the basic protocol described here is efficient and has proven effective in a large number of hESC and hiPSC lines, optimization may be required for specific cell lines.

A. Pluripotent cells are cultured on an E-Cad-Fc substrate before being transferred to Matrigel. Cells are then sequentially exposed to factors with known roles in controlling hepatocyte formation during embryogenesis. Images on right show phase contrast micrographs of cells at each stage of the differentiation procedure. B. Micrograph showing the distribution of pluripotent stem cell colonies on E-cad-Fc immediately before plating for differentiation. C. Higher resolution micrograph of a single colony of pluripotent stem cells immediately before plating for differentiation. D. Micrograph showing distribution of pluripotent stem cells after plating on Matrigel and immediately before initiating differentiation. Scale bar = 50 μm.
2. The Protocol
2.1. A. Transfer of pluripotent cells to feeder–free conditions
It is critical to maintain very high quality stocks of hESCs or hiPSCs, which means paying very close attention to routine culture conditions. If the quality of the starting cell population drops the efficiency of differentiation plummets! For each line we are working with we maintain a stock on DR4 (Tg(DR4)1Jae/J) mitotically inactivated primary embryonic feeder cells (MEFs) that are manually passaged and all differentiated cells are removed from the cultures. We closely follow the rate of cell division which should be slow. If the cells start proliferating more rapidly and passage of cultures becomes more frequent they should be discarded. To maintain the hESC/hiPSCs in an optimal condition that is compatible with efficient differentiation we use either mTeSR1 or MEF-conditioned medium. Finally all culture of hESCs or hiPSCs is performed under low (4%) O2 conditions that has been shown by others to inhibit differentiation (Ezashi et al., 2005).
While the differentiation of the hESCs or hiPSCs toward hepatocytes can be initiated from cells maintained on either MEFs or Matrigel with relative success, we have found that the reproducibility of differentiations is markedly enhanced by maintaining the pluripotent cells on non tissue culture treated suspension culture dishes (see supplemental protocol for specific information on dishes) coated with a fusion protein consisting of the E-cadherin extracellular domain and the IgG Fc domain (E-cad-Fc) (Nagaoka et al., 2010). This substrate offers several advantages over Matrigel as we have explained elsewhere (Nagaoka et al., 2010). For example, E-cad-Fc is a recombinant protein that selectively binds cells that express E-cadherin on the cell surface, including human pluripotent stem cells. As hESC/hiPSC differentiate the cells tend to down regulate expression of E-Cadherin and as a consequence differentiated cells generally fail to adhere to the E-cad-Fc substrate. Culture on this matrix, therefore, inherently selects for a pluripotent phenotype and maintains the cells in a relatively homogenous state. The matrix also allows gentle manipulation of the stem cells because passaging uses simple enzyme-free buffers that maintain cell surface proteins and ensure high cell viability. Although E-cad-Fc can be produced in the laboratory (Nagaoka and Duncan, 2012), a similar matrix called StemAdhere can be obtained commercially (STEMCell Technologies, Inc) ( https://www.stemcell.com/en/Products/All-Products/StemAdhere-Defined-Matrix-for-hPSC.aspx).
2.1.1. A.1. Culture of pluripotent cells using defined media
While MEF-conditioned medium can be used to maintain pluripotent cells, mTeSR1 (STEMCell Technologies, Inc), which is compatible with culture on E-cad-Fc, has proven to be particularly compatible with the hepatocyte differentiation protocol. The use of mTeSR1 has the additional advantage of avoiding the variability often associated with preparations of MEFs and greatly simplifies maintaining high quality cultures of hESC/hiPSCs. Although the purchase of defined culture media can be prohibitively expensive, clearly described protocols for the production of cost-effective mTeSR1 have been published by others that are relatively easily incorporated into the laboratory (Ludwig et al., 2006b; Ludwig et al., 2006a).
2.1.2. A.2. Culture of pluripotent cells using MEF–conditioned hESC medium
-
Plate 1 vial (5 × 106 cells) of DR4 mitotically inactivated feeder fibroblasts on a 100 mm plate in hESC medium and leave overnight at 37°C 5%CO2 to seed.
-
Next day, after cells have attached, remove medium and replace with 10–13 ml of fresh hESC medium (without FGF) and leave overnight.
-
Collect this media (optional - filter through 0.22 μM syringe filter) to use as ‘MEF–conditioned medium’ for ESC culture.
-
Continue to culture and collect medium daily as need dictates.
Note: A plate of MEFs is good for 5–7 days before new MEFs should be thawed. Immediately before use in hESC/hiPSC culture MEF-conditioned medium should be supplemented with 40 ng/ml zebrafish bFGF (zbFGF prepared as described by Ludwig et al (Ludwig et al., 2006a)) or 4 ng/ml FGF2 (Invitrogen).
2.1.3. A.3. Initial transfer of hESC/iPCs to E-cad-Fc
-
Coat 3-wells of a 6-well plate with E-cad-Fc (see supporting protocol below) for every p60 of hESC/iPSCs that need passaged.
Note: The cells may take a few passages on the E-cad-Fc surface to adapt; however, the cells should soon show growth characteristics that are very similar to those of cells grown on Matrigel (for details see (Nagaoka et al., 2010)).
-
Remove medium from the hESC/hiPSCs and replace with 3 ml of fresh MEF-conditioned hESC medium/mTeSR1.
-
Collect undifferentiated hESCs/hiPSCs by manual dissection of the colonies using a 27G needle and displace them with a sterile pipette tip. Fragments of colonies are now floating in the fresh medium within the dish.
-
Transfer the medium (∼3 ml) containing the dissected colonies to a sterile 15 ml conical tube. Add an additional 1.5 ml of fresh MEF-conditioned hESC medium/mTeSR1 and transfer 1.5 ml of cells to each of the 3-wells coated with E-cad-Fc (from 1). This should result in approximately 20–35 colony fragments being transferred per well.
-
Culture cells with daily medium changes for 3–5 days in MEF-conditioned hESC medium/mTeSR1 at 37°C 4%O2/5%CO2.
Note: Plated colony fragments commonly take a few days to flatten out on the surface of the matrix. Also, during the initial transfer to E-cad-Fc, larger colony fragments appear to attach with higher efficiency than small fragments that often fail to adhere. It is therefore important to consider this when manually dissecting the colonies.
2.1.4. A.4. Maintenance of hESC/iPSCs on E-cad-Fc
The working hESC/hiPSC stocks for differentiation are maintained on an E-cad-Fc fusion substrate on non-treated suspension culture dishes and passaged before the cells reach 50% confluence. Do not let the cells become confluent, it is crucial that they are kept as relatively small clusters that resemble the morphology of well defined colonies grown on MEFs (Figure 1B-D).
-
Wash plate to which hESC/hiPSCs are attached with sterile PBS. Remove wash liquid. Add sufficient Versene/0.02% EDTA (Lonza) or Cell Dissociation Buffer (Invitrogen) to each plate and incubate at room temp for no longer than 2 minutes. As soon as the cells appear to detach add 2–3 volumes of culture medium. Remove the cells by carefully pipetting with a 5 ml pipet.
Note: It is important to add media containing Ca++ as soon as cells begin to detach to reduce denaturation of cell surface E-cadherin and maintain high cell viablity.
-
Transfer the cells to a conical tube and collect cells by centrifugation at ∼100–200 g then resuspend in MEF-conditioned hESC medium/mTeSR1. Transfer cells to a suspension culture dish coated with E-cad-Fc (see supplemental protocol).
-
Culture cells with daily medium changes in MEF-conditioned hESC medium/mTeSR1 at 37°C 4%O2/5%CO2. A sub-confluent 100 mm provides between 1.0 – 2.5 × 106 cells. At this stage cells can be expanded to the desired amount by serial passage; however, again it is crucial to keep the cells sub–confluent.
Note: Over 95% of cells at this stage should be Tra1-60 and Tra1-81 positive if examined by FACS.
2.2. B. Preparation of cells for differentiation
To initiate differentiation, cells are harvested from the E-cad-Fc plates as small cell clusters from a plate that contains dividing cells. Do not use hESC/hiPSC cultures that show morphological evidence of differentiation or cover more than 50% of the dish. The cells are transferred to a Matrigel–coated plate at a concentration that allows the cells to become close to confluent after no longer than an overnight incubation. The exact number of cells plated needs to be determined empirically and varies from cell line to cell line. As a rule of thumb, a 100 mm plate that is estimated by visual inspection to be ∼50% confluent (Figure 1B, C) provides sufficient cells to seed 2 (35 mm) wells of a 6–well plate (Figure 1D).
-
Briefly rinse each plate of hESC/hiPSCs with sterile Ca++/Mg++ free PBS. If collecting cells from a 100 mm culture dish add 3 ml of Accutase (scale down as necessary) and incubate at room temp for ∼ 2 minutes. This step is critical. After two minutes, examine the plate and tilt to determine if cells are lifting from the E-cad-Fc surface. Once cells are observed sloughing off of the plate immediately neutralize with 1–2 volumes of medium.
Note: For some cell lines enzyme free Cell Dissociation Buffer (Invitrogen) can be used instead of Accutase. This should be determined empirically. Wash cells with 2–3 ml PBS, remove and add 2 ml Cell Dissociation Buffer and incubate for 2–3 mins. After cells begin to detach collect in medium and follow step 2.
-
Harvest cells using a serological pipet and transfer to a 15 ml conical tube. Collect by centrifugation at ∼100–200 g then resuspend in 1 ml of MEF-conditioned hESC medium/mTeSR1.
-
Transfer cells to the appropriate number of wells in a 6–well plate coated with Matrigel in MEF–conditioned hESC medium/mTeSR1 (see protocol below). Culture overnight at 37°C 4%O2/5%CO2.
2.3. C. Differentiation of pluripotent cells to hepatocytes
After overnight culture it is important that the cells should be ∼90% confluent before beginning the differentiation protocol. Do not culture for longer than 24 h before initiating differentiation, because this results in a dramatic decrease in efficiency of differentiation.
-
(Differentiation Day 1) Remove medium and replace with RPMI/B-27(minus Insulin) (Invitrogen/Gibco) supplemented with 100 ng/ml Activin A (R&D systems), 10 ng/ml BMP4 (Peprotech) and 20 ng/ml FGF2 (Invitrogen). Change medium daily and culture for 2–days at 37°C in ambient O2/5%CO2.
Note: We have observed that there appears to be substantial variation in efficiency of differentiation between specific lots of B27. In response to similar concerns raised by scientists culturing neuronal cells an alternative supplement called NS21 was developed (Chen et al., 2008). We have found that NS21 is a good substitute for B27 that can easily be generated in the laboratory following published procedures (Chen et al., 2008). Moreover, because the formulation of NS21 is in the public domain it is relatively easy to optimize.
-
(Differentiation Day 3–5) Change culture medium to RPMI/B-27(minus Insulin) supplemented with 100 ng/ml Activin A and continue to culture for an additional 3–days at 37°C in ambient O2/5%CO2 with daily medium changes.
Note: After 5-days of culture 80–90% of the cells should express endodermal markers, FOXA2 (Figure 2A), SOX17 (Figure 2B), GATA4 (Figure 3A), and CXCR4, and the presence of HNF4a and OCT4 expressing cells should be minimal.
 Figure 2.
Figure 2.Expression of characteristic definitive endoderm proteins can be detected in 80–90% of cells following treatment of pluripotent stem cells with Activin A. Micrographs show the presence of endodermal proteins (A) FOXA2 or (B) SOX17 (green), or pluripotency marker protein (B, E) OCT4 (red) detected by immunocytochemistry in the nuclei of cells after 5–days of differentiation. Staining with DAPI (C, F) identified cell nuclei. Scale bars = 50 μm.
 Figure 3.
Figure 3.Characteristic markers can be identified at each stage of differentiation. Immunocytochemistry was used to identify A) GATA4, B) HNF4A, C) AFP, and D) ALB in cells following 5, 10, 15 and 20 days of differentiation, respectively. Total cell population was revealed by DAPI staining of nuclei (E-F). Scale bar = 50 μm. I) Bar graph showing the level of specific hepatic mRNAs found in cells that have completed the differentiation protocol determined by qRT-PCR. Levels of mRNA are presented as a fraction of those found fresh primary human hepatocytes.
-
(Differentiation Day 6) Change medium to RPMI/B27 (complete with Insulin) supplemented with 20 ng/ml BMP-4 and 10 ng/ml FGF2. Culture for 5–days at 37°C in 4%O2/5%CO2 and replace medium daily for a total of 5–days.
Note: By the end of day 10, following BMP4/FGF2 treatment, cells should uniformly express FOXA2 and HNF4a (Figure 3B) and levels of SOX17 and GATA4 should have diminished.
-
(Differentiation Day 11) Change medium to RPMI/B27(complete with Insulin) supplemented with 20 ng/ml Hepatocyte Growth Factor (HGF) (Peprotech). Culture for 5–days at 37°C in 4%O2/5%CO2 and replace medium daily.
Note: Following culture of cells in HGF ∼80% of cells should express AFP (Figure 3C) with low to undetectable expression of ALB. The cells should also have a characteristic cuboidal morphology and a large cytoplasmic to nuclear ratio. Fat droplets should also be detectable within the cells.
-
(Differentiation Day 16) Change medium to Clonetics® HCM™ Hepatocyte Culture Medium (Lonza CC-3198) containing ‘Singlequots’ (see HCM medium below) omitting the EGF from the Lonza HCM bullet kit. In addition to the kit supplements, add 20 ng/ml Oncostatin-M (R&D Systems). Culture for 5–days at 37°C in ambient O2/5%CO2 and replace medium daily.
Note: After 5–days of culture (Differentiation Day 20) 70–90% of cells should express characteristic hepatic markers including Albumin (Figure 3D). Several markers that are characteristic of the differentiated state can be detected by qRT-PCR (Figure 3I) or FACS including ASGPR1, ALB, APOH, APOA1, APOB, FGA, AGT, HGD, TF, and GLUT2. For an extensive list of markers and gene array analyses of the differentiation time course see (Delaforest et al., 2011). In addition cell junction proteins are detectable at the cell surface such as E-cadherin, Claudin1, and Occludin and the cells are capable of typical hepatic functions including glycogen synthesis, indocyanine green metabolism, Albumin secretion, and LDL-C uptake as well as infection with hepatitis C virus (Schwartz et al., 2012).
3. Conclusion
The reproducibility of the protocol relies heavily on the quality and pluripotency of the starting hESC or hiPSC population. For this reason a focus on maintaining high quality hESCs and hiPSCs is of paramount importance – poor cells result in poor differentiations! For this reason cultures of hESC and hiPSCs should be closely followed and routinely tested for expression of pluripotency markers, such as SSEA4, TRA1-60, and TRA1-81, by FACS and particular attention should be paid to the rate of cell proliferation. If, in a given culture, such parameters deviate from normal the cells should be discarded and new cultures reestablished from earlier passages with confirmed karyotypes and documented growth characteristics.
Importantly, we have also found that commercial additives including growth factors, supplements, and extracellular matrices exhibit substantial variation in quality between batches. Most commercial suppliers do not provide the specific activity of a given growth factor between production lots and, of course, differences in the activity of a given growth factor can have a considerable impact on the efficiency of differentiation and maintenance of pluripotency. Using reporter cell lines to determine specific activity of purchased growth factors can minimize such variability. When possible we have elected to purify our own growth factors and cell matrices following published procedures, which has the added benefit of saving tens of thousands of dollars in cell culture costs (Ludwig et al., 2006a; Zou and Sun, 2004; Nagaoka and Duncan, 2012).
Although this protocol is robust and routinely yields high quality cells, it is important to bear in mind that the resulting cells do not fully recapitulate all functional aspects of a primary human hepatocyte. As we have discussed previously (Si-Tayeb et al., 2010a), and as appears to be the case for perhaps all hepatocyte differentiation protocols described to date, the level of key functional proteins including CYP450 enzymes are below that found in fresh human hepatocytes. In addition, in contrast to adult liver cells, hepatocytes derived from human pluripotent stem cells retain expression of AFP, which is consistent with the belief that the cells retain fetal hepatocyte characteristics.

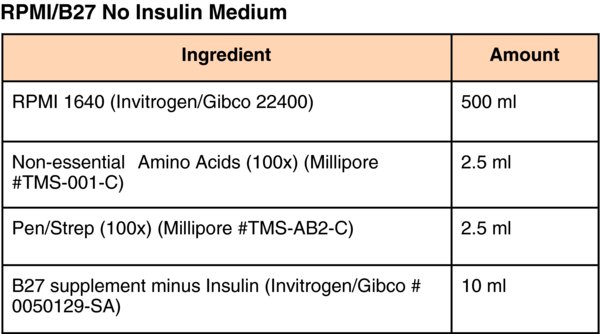


5. Supplemental protocols
5.1. A. Coating plates with E-cad-Fc
-
Thaw an aliquot of stock E-cad-Fc (500 μg/ml) from storage at –80°C. Dilute E-cad-Fc in PBS+ (PBS w/calcium chloride/magnesium chloride, DPBS Gibco 14040) to 10 μg/ml.
-
Coat the base of each plate with the appropriate volume of E-Cad-Fc. It is essential that non-treated polystyrene cell culture plates are used.
-
100 mm plate (Suspension Culture Plates, VWR #25382-456) = 5.0 ml
-
60 mm plate (Suspension Culture Plates, VWR #25382-452) = 2.0 ml
-
6-well plate (Cellstar Non-TC coated, BioExpress T-3026-4) = 1.0 ml/well
-
24-well plate (BD Falcon Non-Treated, VWR #08-772-51) = 300 μl/well
-
96-well plate (Corning Costar Assay Plate, VWR #25381-056) = 50 μl/well
Note: The brand of the tissue culture dishes used can have an impact on the quality of pluripotent cells and in the efficiency of cell attachment to E-Cad-Fc. Also suspension culture plates are hydrophobic and so it is necessary to quite vigorously rock the plates in order to evenly coat the surface with E-cad-Fc.
-
-
Incubate plates at 37°C for a minimum of 1 hr.
-
When ready to plate cells, remove coating liquid and optionally wash 1X with PBS+.
Note: If using StemAdhere follow manufacturers instructions.
5.2. B. Coating plates with Matrigel
-
Prepare a tube with 10 ml of pure DMEM/F12 medium (no serum, antibiotic or other additives) and chill on ice.
-
Retrieve a 250 μl aliquot of Matrigel (2 mg/ml) (GFR Matrigel, BD Bioscience or Geltrex hESC-qualified Reduced Growth Factor Basement Membrane Matrix, Invitrogen) from storage at –80°C. Use 250 μl Matrigel for each 10 ml of medium.
-
Resuspend the frozen Matrigel with the cold medium but do not allow it to come to room temperature or it will solidify.
-
When the aliquot is dissolved in the cold medium add appropriate volume to each plate/well.
-
100 mm Tissue Culture Dish (Corning Tissue Culture Dish, Bioexpress T-2877-100) = 4.0 ml
-
60 mm Tissue Culture Dish (Corning Tissue Culture Dish, Bioexpress T-2877-60) = 2.0 ml
-
6 well Plates (Corning Costar Tissue Culture Plate, Bioexpress T2989-6) = 1.5 ml
-
12 well Plates: Corning Costar Tissue Culture Plate, Bioexpress T2989-12 = 800 μl
-
24 well Plates: Corning Costar Tissue Culture Plate, Bioexpress T2989-24 = 400 μl
-
96 well Plates: Corning Costar Tissue Culture Plate, Bioexpress T2989-96 = 50 μl
Note: The brand of the tissue culture dishes used has an impact on the quality of differentiations.
-
-
Leave plates at 37°C for 1 hr.
-
Aspirate the medium and either plate cells or cover plate with hESC media and keep it in the incubator until needed.
Acknowledgements
The authors would like to thank Karim Si-Tayeb, Fallon Noto, Michele Battle, and all previous members of the Duncan lab for their valuable advice. Work described here was supported by gifts from the Marcus Family, Phoebe R. and John D. Lewis Foundation, the Sophia Wolf Quadracci Memorial Fund, the Dr. James Guhl Memorial Fund and the Advancing a Healthier Wisconsin Fund, as well as NIH grants DK55743, DK087377, HL107434 and HL094857 to S.A.D. J.C. was supported by JDRF post-doctoral fellowship 3-2010-497 and M.C. is supported by F30 DK091994.
